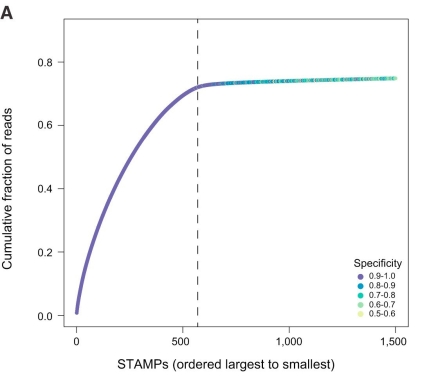
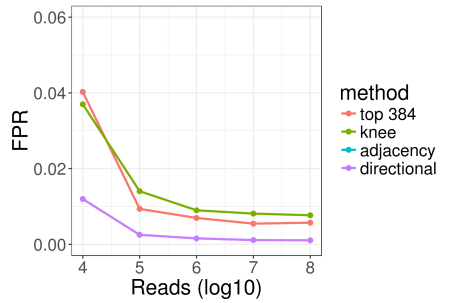
This post starts where we left off in the first part. If you haven’t already done so, I’d recommend reading the first part now.
I hadn’t planned on writing another post but no sooner had I finished the first part of this blog post than I discovered that the Pachter group have developed a tool to perform the same task (sircel, pronounced ‘circle’). This appears to be in the early stages of development but there’s a bioAxiv manuscript out which happens to cover the same input data as I discussed in the previous post (thanks to Avi Srivastava for bringing this to my attention).
In the discussion with Avi in the comments of the previous post I also realised that I had failed to clarify that only base substitutions will be detectable when using our network methods. As it happens, one of the motivations for the development of sircel appears to be handling INDELs which they claim can be present in >25% of dropseq barcodes. I can’t see where they get this number from (In the manuscript they reference the original Macosko et al paper but I can’t see this 25% figure anywhere) so I’m assuming for now that this is based on additional analyses they’ve performed. This seems like an alarmingly high figure to me given that the rate of INDELs is ~1 order of magnitude less frequent than substituions (see Fig. 2/Table 2 here) and even at the start of the read where the cell barcode resides, the per-base rate of INDELs does not usually exceed 0.001. I would also assume that cell barcodes with INDELs would have far fewer reads and so drop below the threshold using the ‘knee’ method. Hence, while some cell barcodes may be discarded due to INDELs, I would be surprised if a INDEL-containing barcode was erroneously included in the accepted cell barcodes.
How common are INDELs?
Before I tested out sircel, I wanted to independently assess what proportion of cell barcodes might have a substitution or INDEL. To do this, I returned to the drop-seq data from the last post (GSE66693). Previously, I showed the distribution of reads per cell barcode separated into 4 bins, where bin 4 represents the accepted barcodes above the knee threshold and bins 1-3 are largely populated by different types of erroneous barcodes (reproduced below as Figure 1). I compared each barcode to all barcodes in bin 4 and looked for a match allowing for one error (substitution, insertion or deletion). The results are tabulated below. These figures are an upper estimate of the number of barcodes/reads harbouring an error relative to the true cell barcodes since each barcode may contribute to all three error types if at least one match to a bin 4 barcode exists with each error type. We can see that ~1.5% of cell barcodes may contain an INDEL, far less than the 25% suggested in the sircel manuscript. Furthermore, when we present the figures according to the number of reads, it appears around 3% of reads have a cell barcode with an INDEL, whilst 7% have a substitution. Thus, we can be quite confident that we’re not losing lots of data if we don’t error correct cell barcodes or just correct substitution errors.

Figure 1. Distribution of reads per cell barcode (log 10) from first 50M reads, separated into 4 bins. Bin 4 represents the ‘true’ barcodes.

Table 1: Columns 1 & 2: The number and fraction of cell barcodes which match a bin 4 barcode allowing one error. Columns 3 & 4: The number and fraction of reads with a cell barcodes which match a bin 4 barcode allowing one error.
Are INDEL-containing barcodes passing the knee threshold?
Next, I want to investigate whether the INDEL barcodes pass the knee threshold and become ‘accepted’. I therefore split the above data according to bin and focused on bin 4. As you can see in Figure 2 below, there are indeed a small number of bin 4 barcodes which are a single error away from another bin 4 barcodes. Our null expectations (from 100 000 randomly generated barcodes) for the percentage of subsitution, deletion and insertion matches by chance are 0.14, 0.11 and 0.13%, respectively. In contrast, amongst the 575 bin 4 barcodes, there are 6 barcodes (1.0%) which match at least one other bin 4 barcode with one error. 4/6 of these barcodes have a match with an INDEL and all have relatively low counts suggesting these are indeed errors. It seems I may have been wrong to assume a INDEL-containing barcodes will not pass the knee threshold. The results for bins 1-3 minor my previous analysis using edit-distance and suggest the bin3 is mainly made up of error-containing barcodes whilst bins 1 & 2 appear to originate from amplification of ambient RNA in empty droplets.
 Figure 2. The frequency of cell barcodes in each bin which are one error away from at least one of the bin 4 barcodes. Random = 100 000 randomly generated barcodes
Figure 2. The frequency of cell barcodes in each bin which are one error away from at least one of the bin 4 barcodes. Random = 100 000 randomly generated barcodes
How does sircle compare to the knee method?
So, INDELs are observed in this drop-Seq data, although perhaps not 25% of all cell barcodes, and INDEL-containing barcodes can pass the hard knee threshold for acceptance. Now on to sircel. As you would expect from the Pachter group, sircel introduces a novel, elegant solution to detect substitutions and INDELs in barcodes. Rather than using edit-distance or fuzzy matching, they ‘circularise’ the barcodes and form a de Bruijn graph using the kmers from the circularised barcodes. The circularisation ensures that even with a large kmer size, barcodes containing an error will share at least a portion of their path through the de Bruijn graph with the cognate error-free barcodes. The edges are weighted according to the number of times each edge is observed and each circular path is given a score according to the minimal weight of all its edges. This score is used to filter the paths to select the high weight, correct paths and incorrect paths are merged to the correct paths. Notably, the thresholding is done using a method analogous to the knee method, whereby the threshold is set as the local maxima of the smoothed first derivative from the cumulative distribution of path scores. The neat thing about the sircel method is that it enables simultaneous identification of this inflection point to identify the threshold for true barcodes, and error correction to the true barcodes. To compare the results for sircel and my knee method, I ran sircel on the GSE66693 drop-seq sample where the authors had suggested there were ~570 true cell barcodes. Previously, using the first 50M reads I had estimated that there were 573 true barcodes using the knee method. As per the sircel manuscript recommendation, I used only 1M reads to run sircel, which yielded 567 true barcodes. This is slightly lower than the number quoted in the manuscript for 1M reads (582) but perhaps this is due to more recent updates? The methods agree on 563 cell barcodes but what about the cell barcodes where they agree. 4/10 out of the knee method-specific cell barcodes are a single error away from another barcode above the threshold, suggesting these may in fact be errors. The remaining 6 barcodes all have very low counts. Likewise, the 4 sircel-specific cell barcodes fall just below the knee threshold (6170-9576 reads per barcode; threshold = 9960). For these 10 barcodes, the difference appears to be due to differences in the number of reads sampled. I wanted to check this by using 50M reads with sircel but the run-time becomes excessively long. Unfortunately, going to other way and using just 1M reads with the knee method yields unstable estimates and since it takes <6 minutes to run the knee method with 50M reads we would never recommend using the knee method with just 1M reads. So in essence, the major difference between the knee method and sircel is that sircel effectively corrects the small number of error barcodes above the threshold.
Can we improve the knee method?
It does look like sircel does a good job of removing error barcodes but we could easily replicate this performance by performing an INDEL-aware error detection step after the knee threshold. During the development of UMI-tools we considered detecting INDELs as well as substitutions but decided against it because: a) as mentioned at the top, the rate of INDELs is usually ~1 order of magnitude lower than substituions (that this doesn’t appear to hold true for cell barcodes), and b) UMI-tools groups reads together by their alignment position so the current framework does not allow the detection of INDELs. However, when we’re dealing with cell barcodes, we consider all barcodes at once and hence, it’s much easier to identify INDELs. For our network methods, we can simply replace the edit-distance threshold with a fuzzy regex, allowing one error (substitution, insertion or deletion). The fuzzy regex matching is ~30-fold slower than our cythonised edit-distance function but this isn’t an issue here since we only need to build the network from the cell barcodes which pass the knee threshold. From the network we can identify the error barcodes and either correct these to the correct barcode or else discard them. This will now be the default method for UMI-tools extract with the option to discard, correct or ignore potential barcode errors above the knee threshold. We will also include the option to correct barcodes which fall below the threshold.
A final thought: To correct or not?
The cost of incorrectly merging two cell barcodes through cell barcode error correction could be drastic, e.g appearing to create cells ‘transitioning’ between discrete states. On the other hand, discarding reads from error barcodes rather than correcting them reduces the sequencing depth and hence increases the noise. In the drop-seq data above 55.7% of reads are retained without error correction with a further 9.2% of reads potentially correctable. If all barcodes above the threshold and within one error of another barcode above the threshold were removed, this would only mean the loss of 0.3% of reads. Given the potential dangers of incorrect error correction, it may be prudent in some cases to discard all cell barcodes with errors and work with 55.4% of the input reads rather than correcting errors in order to retain 64.9% or reads